Identification of differentially expressed peptides in high-throughput proteomics data
Abstract
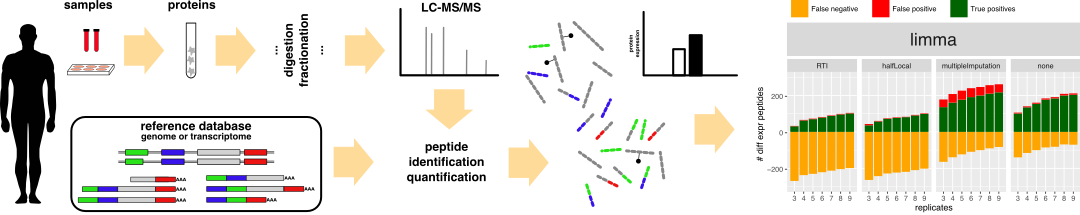
With the advent of high-throughput proteomics, the type and amount of data pose a significant challenge to statistical approaches used to validate current quantitative analysis. Whereas many studies focus on the analysis at the protein level, the analysis of peptide-level data provides insight into changes at the sub-protein level, including splice variants, isoforms and a range of post-translational modifications. Statistical evaluation of liquid chromatography–mass spectrometry/mass spectrometry peptide-based label-free differential data is most commonly performed using a t-test or analysis of variance, often after the application of data imputation to reduce the number of missing values. In high-throughput proteomics, statistical analysis methods and imputation techniques are difficult to evaluate, given the lack of gold standard data sets. Here, we use experimental and resampled data to evaluate the performance of four statistical analysis methods and the added value of imputation, for different numbers of biological replicates. We find that three or four replicates are the minimum requirement for high-throughput data analysis and confident assignment of significant changes. Data imputation does increase sensitivity in some cases, but leads to a much higher actual false discovery rate. Additionally, we find that empirical Bayes method (limma) achieves the highest sensitivity, and we thus recommend its use for performing differential expression analysis at the peptide level.